Fine-tuning a Generative Model¶
Fine-tune ESM3 or ESMC with LoRA on a family of protein sequences, optionally conditioned on per-sequence structures for inverse folding.
Overview¶
Two modes:
- Sequence-only MLM: mask random positions, predict them from surrounding sequence context
- Inverse folding: mask all sequence positions, predict them from structure alone
Sequence-only fine-tuning¶
Train the model to better predict masked amino acids within a protein family. Uses ProteinDataset with uniform_mask_noise to randomly mask positions.
Setup¶
from proteingen.data import ProteinDataset, uniform_mask_noise, uniform_time
from proteingen.models.esm import ESM3
from torch.utils.data import DataLoader
# Load sequences (see MSA → Dataset workflow)
dataset = ProteinDataset(sequences=my_sequences)
# Load model with LoRA
model = ESM3("esm3-open")
model.apply_lora(r=4, lora_alpha=8)
model.to("cuda")
# Freeze everything except LoRA
for name, p in model.named_parameters():
p.requires_grad = "lora_" in name
# Build collator — noise_fn and time_sampler are required
noise_fn = uniform_mask_noise(model.tokenizer)
collate_fn = dataset.collator(model, noise_fn=noise_fn, time_sampler=uniform_time)
loader = DataLoader(dataset, batch_size=4, shuffle=True, collate_fn=collate_fn)
Training loop¶
The collator returns input_ids (noised) and target_ids (clean). Compute loss only on masked positions:
for batch in loader:
input_ids = batch["input_ids"].to(device)
target_ids = batch["target_ids"].to(device)
raw = model(input_ids)
logits = model.format_raw_to_logits(raw, input_ids)
# Loss only where masking changed the token
masked = input_ids != target_ids
loss = F.cross_entropy(logits.float()[masked], target_ids[masked])
optimizer.zero_grad()
loss.backward()
torch.nn.utils.clip_grad_norm_(
[p for p in model.parameters() if p.requires_grad], 1.0
)
optimizer.step()
Use AMP on GPU
ESM3 logits can overflow in fp32, producing inf loss. Always use torch.amp.autocast(device_type="cuda", dtype=torch.bfloat16) for GPU training.
Example results: EphB1 kinase domain¶
Fine-tuning ESM3 (LoRA r=4) on ~10k EphB1 homologs from a UniRef MSA:
| Epoch | Loss (NLL) | Perplexity |
|---|---|---|
| 1 | 1.80 | 6.04 |
| 2 | 1.67 | 5.33 |
| 3 | 1.64 | 5.16 |
| 4 | 1.61 | 5.00 |
| 5 | 1.60 | 4.96 |
Loss is computed only on masked positions — perplexity reflects actual prediction difficulty, not diluted by trivially-correct unmasked positions.
Full script: examples/finetune_esm3/finetune_esm3_ephb1.py
Inverse folding (structure-conditioned)¶
Train the model to predict sequence from structure. Each sequence has its own predicted structure (from AF3), so variable-length sequences are handled naturally.
Setup¶
import torch
from torch.utils.data import DataLoader
from proteingen.models.esm import ESM3
# Load pre-computed structures (see MSA → Dataset workflow)
data = torch.load("ephb1_structures.pt", weights_only=False)
# Custom dataset for (sequence, structure) pairs
dataset = InverseFoldingDataset(
sequences=data["sequences"],
structure_tokens=data["structure_tokens"],
coordinates=data["coordinates"],
)
model = ESM3("esm3-open")
model.apply_lora(r=4, lora_alpha=8)
model.to("cuda")
Training loop¶
For inverse folding, mask all non-special positions and condition on structure:
for batch in loader:
input_ids = batch["input_ids"].to(device) # fully masked
target_ids = batch["target_ids"].to(device) # true sequence
struct_tokens = batch["structure_tokens"].to(device)
coords = batch["coordinates"].to(device)
maskable = batch["maskable"] # non-special positions
raw = model(input_ids, structure_tokens=struct_tokens, coordinates=coords)
logits = model.format_raw_to_logits(
raw, input_ids, structure_tokens=struct_tokens, coordinates=coords
)
loss = F.cross_entropy(logits.float()[maskable], target_ids[maskable])
loss.backward()
optimizer.step()
The collator pads both sequences and structures to the batch max length, using the tokenizer's pad token for sequences and STRUCTURE_PAD_TOKEN for structures.
Example results: EphB1 inverse folding¶
Fine-tuning ESM3 (LoRA r=4) on ~9.2k EphB1 homologs with AF3-predicted structures:
| Epoch | Loss | PPL | Struct log p (t=0) | Seq-only log p (t=0) |
|---|---|---|---|---|
| 0 (pretrained) | — | — | -2.075 | -2.955 |
| 1 | 1.034 | 2.81 | -1.026 | -2.953 |
| 2 | 0.761 | 2.14 | -0.911 | -2.952 |
| 3 | 0.673 | 1.96 | -0.871 | -2.952 |
| 4 | 0.616 | 1.85 | -0.832 | -2.952 |
| 5 | 0.572 | 1.77 | -0.798 | -2.953 |
Key observations:
- Structure-conditioned log probability improved dramatically: -2.075 → -0.798 (model learns to use structure for sequence prediction)
- Sequence-only log probability stayed flat at ~-2.95 (model isn't memorizing sequences — it genuinely relies on structure)
- Final perplexity of 1.77 means the model predicts the correct amino acid ~56% of the time from structure alone
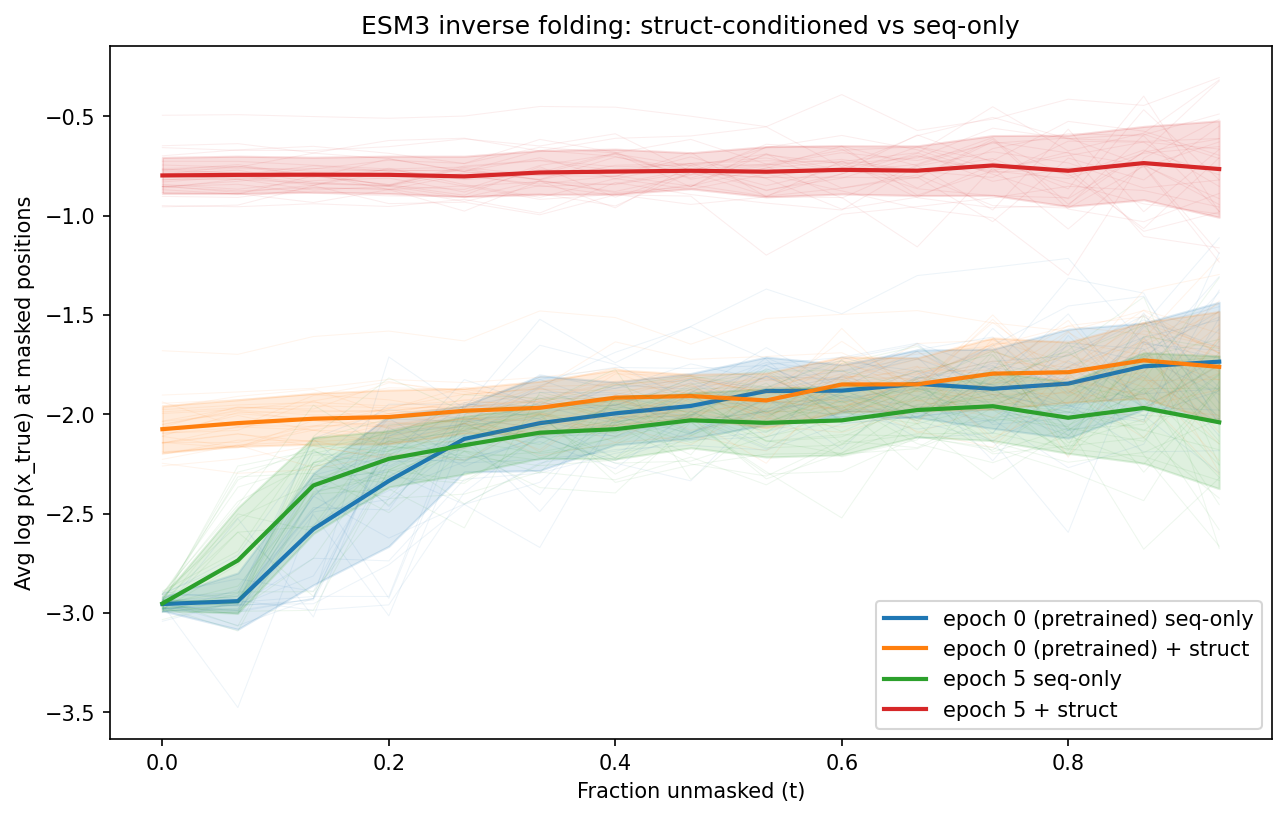
Likelihood curves comparing structure-conditioned (top) vs sequence-only (bottom) prediction. The fine-tuned model with structure (red) achieves uniformly high log probabilities regardless of how much sequence is masked.
Full script: examples/finetune_esm3/finetune_inverse_folding.py
Saving and loading checkpoints¶
# Save LoRA adapter
model.save("checkpoints/my_model")
# Load later
model = ESM3("esm3-open")
model.load("checkpoints/my_model") # applies LoRA + loads adapter weights
model.to("cuda")
The checkpoint saves only the LoRA adapter weights (~19MB for r=4), not the full model.
wandb logging¶
Both example scripts log to wandb. Key metrics:
train/loss,train/ppl— per-step training metricsepoch/loss,epoch/ppl— per-epoch averageseval/struct/log_prob_t0— structure-conditioned log prob at full maskingeval/seq_only/log_prob_t0— sequence-only log prob at full maskingeval/likelihood_curves— comparison plot uploaded as image